Difference between revisions of "File:Fig1 Reid BMCInformatics2014 15.jpg"
Shawndouglas (talk | contribs) (Added summary.) |
Shawndouglas (talk | contribs) m (→Summary: Desc) |
||
| Line 1: | Line 1: | ||
==Summary== | ==Summary== | ||
{{Information | {{Information | ||
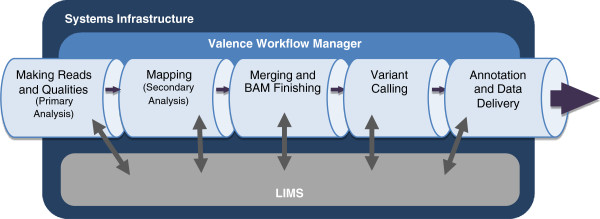
|Description='''Mercury Data Flow.''' 1) Sequencing Instrument raw data is passed to vendor primary analysis software to generate sequence reads and base call confidence values (qualities). 2) Reads and qualities are passed to a mapping tool (BWA) for comparison to a reference genome to determine the placement of reads on the reference (producing a BAM file). 3) Individual sequence event BAMs are merged to make a single sample-level BAM file that then is processed in preparation for variant calling. 4) Atlas-SNP and Atlas-indel are used to identify variants and produce variant files (VCF). 5) Annotation adds biological and functional information to the variant lists and formats them for delivery. | |Description='''Figure 1. Mercury Data Flow.''' 1) Sequencing Instrument raw data is passed to vendor primary analysis software to generate sequence reads and base call confidence values (qualities). 2) Reads and qualities are passed to a mapping tool (BWA) for comparison to a reference genome to determine the placement of reads on the reference (producing a BAM file). 3) Individual sequence event BAMs are merged to make a single sample-level BAM file that then is processed in preparation for variant calling. 4) Atlas-SNP and Atlas-indel are used to identify variants and produce variant files (VCF). 5) Annotation adds biological and functional information to the variant lists and formats them for delivery. | ||
|Source={{cite journal |url=http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-30 |title=Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline |journal=BMC Bioinformatics |author=Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric |volume=15 |pages=30 |year=2014 |doi=10.1186/1471-2105-15-30 |issn=1471-2105}} | |Source={{cite journal |url=http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-30 |title=Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline |journal=BMC Bioinformatics |author=Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric |volume=15 |pages=30 |year=2014 |doi=10.1186/1471-2105-15-30 |issn=1471-2105}} | ||
|Author=Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric | |Author=Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric | ||
Latest revision as of 18:36, 18 December 2015
Summary
| Description |
Figure 1. Mercury Data Flow. 1) Sequencing Instrument raw data is passed to vendor primary analysis software to generate sequence reads and base call confidence values (qualities). 2) Reads and qualities are passed to a mapping tool (BWA) for comparison to a reference genome to determine the placement of reads on the reference (producing a BAM file). 3) Individual sequence event BAMs are merged to make a single sample-level BAM file that then is processed in preparation for variant calling. 4) Atlas-SNP and Atlas-indel are used to identify variants and produce variant files (VCF). 5) Annotation adds biological and functional information to the variant lists and formats them for delivery. |
|---|---|
| Source |
Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric (2014). "Launching genomics into the cloud: Deployment of Mercury, a next generation sequence analysis pipeline". BMC Bioinformatics 15: 30. doi:10.1186/1471-2105-15-30. ISSN 1471-2105. http://bmcbioinformatics.biomedcentral.com/articles/10.1186/1471-2105-15-30. |
| Date |
2014 |
| Author |
Reid, Jeffrey G.; Carroll, Andrew; Veeraraghavan, Narayanan; Dahdouli, Mahmoud; Sundquist, Andreas; English, Adam; Bainbridge, Matthew; White, Simon; Salerno, William; Buhay, Christian; Yu, Fuli; Muzny, Donna; Daly, Richard; Duyk, Geoff; Gibbs, Richard A. Boerwinkle, Eric |
| Permission (Reusing this file) |
|
| Other versions |
Licensing
|
|
This work is licensed under the Creative Commons Attribution 2.0 License. |
File history
Click on a date/time to view the file as it appeared at that time.
| Date/Time | Thumbnail | Dimensions | User | Comment | |
|---|---|---|---|---|---|
| current | 18:25, 18 December 2015 | 600 × 219 (32 KB) | Shawndouglas (talk | contribs) |
You cannot overwrite this file.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}